-
Enantiospecific Elongation of Cationic Helicenes by Electrophilic Functionalization at Terminal Ends
R. Duwald, S. Pascal, J. Bosson, S. Grass, C. Besnard, T. Buergi and J. Lacour
Chemistry - A European Journal, 23 (55) (2017), p13596-13601


DOI:10.1002/chem.201703441 | unige:97330 | Abstract | Article HTML | Article PDF | Supporting Info
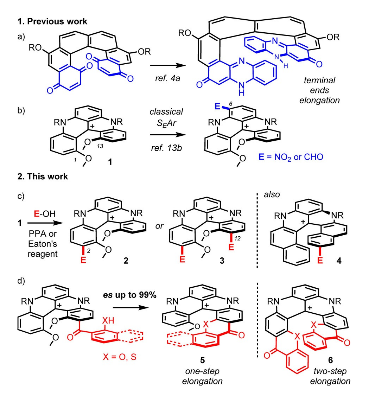
A strategy for late-stage electrophilic functionalizations of cationic helicenes is exposed. Thanks to strongly acidic conditions that permit reversible electrophilic substitutions, regioselective acylations, sulfonylations or alkylations occur at the extremity(ies) of the helical cores. Extended [5] or [6]helicenes can then be generated from cationic [4]helicenes in successive one-pot elongation processes. Retention of configuration and excellent enantiospecificity (up to 99%) are observed for the helicene growth in the enantiopure series.